Antikörper stellen die dominierende Klasse von Proteintherapeutika dar, mit über 160 global lizenzierten Antikörpertherapeutika und einer erwarteten Marktwertsteigerung auf 445 Milliarden US-Dollar in den nächsten fünf Jahren.
Die Entwicklung von Antikörpern erfolgt in der Regel in zwei Phasen: (1) der Entdeckung von Antikörpern, die spezifisch an ein bestimmtes Epitop binden, und (2) der anschließenden Affinitätsreifung und klinischen Optimierung dieser Antikörper.
Aktuell basiert die Identifizierung von epitop-spezifischen Antikörpern auf der Immunisierung von Tieren oder dem Screening von Antikörperbibliotheken, um Kandidatenmoleküle zu ermitteln, die an ein gewünschtes Ziel binden, gefolgt von anschließender Epitope-Kartierung.
Diese Methoden sind zeitaufwändig, arbeitsintensiv und können versagen, Antikörper zu identifizieren, die mit dem therapeutisch relevanten Epitop interagieren.
Die bisherigen Anstrengungen im Bereich des computergestützten Antikörperdesigns konzentrierten sich hauptsächlich auf den zweiten Optimierungsschritt der Antikörperentwicklung, etwa durch das Sampling alternativer nativer CDR-Schleifen zur Verbesserung der Affinitäten oder die Verwendung von Rosetta-Sequenzdesign zur Optimierung der Interaktionsbereiche.
In letzter Zeit wurden struktur- und sequenzbasierte Deep-Learning-Netzwerke trainiert, um neuartige Antikörpersequenzvarianten zu entwerfen, jedoch erfordern diese Methoden einen anfänglichen bindenden Antikörper, von dem aus optimiert werden kann.
Zudem gab es jüngste Fortschritte bei der Antikörperoptimierung mit Deep-Learning-Methoden, die auf Daten trainiert wurden, die durch leistungsstarke neue experimentelle Methoden generiert wurden.
Im Gegensatz dazu existieren jedoch keine computergestützten Methoden zur Durchführung der ersten Phase des Antikörperdesigns (generieren von epitop-spezifischen bindenden Antikörpern), weshalb das de novo-Design von Antikörpern (keine Homologie zu einem bestehenden Antikörper, der dieses Epitop anvisiert) nach wie vor ein ungelöstes Problem darstellt.
Es gab schnelle Fortschritte bei der Gestaltung von Bindungsproteinen (nicht Antikörpern) unter Verwendung von RFdiffusion.
Allerdings sind diese Binder fast ausschließlich auf reguläre Sekundärstruktur-basierte (helikal oder strand) Wechselwirkungen mit dem Ziel-Epitop angewiesen und das ursprüngliche (‘vanilla’) RFdiffusion-Netzwerk ist daher nicht in der Lage, Antikörper de novo zu entwerfen.
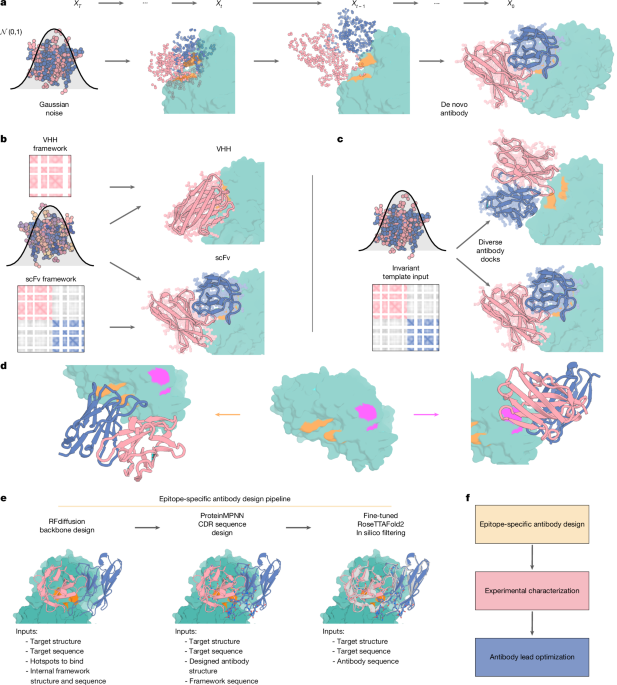
Eine ideale Methode zum Entwerfen von de novo Antikörpern sollte die folgenden Anforderungen erfüllen: (1) das Targeting eines bestimmen Epitops auf einem beliebigen Ziel von Interesse; (2) die Fokussierung der Sampling auf die CDR-Schleifen, während die Framework-Sequenz und -struktur nahe einer benutzerdefinierten, hoch optimierten therapeutischen Antikörper-Framework bleibt; und (3) das Sampling von alternativen Rigid-Body-Platzierungen des entworfenen Antikörpers in Bezug auf das Epitop.
Wir haben die Hypothese aufgestellt, dass eine spezialisierte Version von RFdiffusion, die auf Antikörperstrukturen feinabgestimmt wurde, in der Lage sein sollte, de novo CDR-vermittelte Interfaces zu gestalten, gegeben die Diversität und Qualität der von RFdiffusion gestalteten de novo Interfaces und unter der Annahme, dass die zugrunde liegenden Thermodynamiken der Interfacenbildung gleich sind.
RFdiffusion verwendet die AlphaFold2- und RF2-Frame-Darstellung von Protein-Rückgraten, die aus den Cα-Koordinaten und der N-Cα-C-rigiden Orientierung für jedes Residuum bestehen.
Während des Trainings wird ein Noising-Zeitplan verwendet, der über eine festgelegte Anzahl von ‘Timesteps’ (T) die Protein-Frames in Richtung zufälliger Priorverteilungen korrumpiert.
Die Cα-Koordinaten werden mit dreidimensionalem Gaußrauschen und die Restorientierungen mit Brownian Motion auf SO3 korrumpiert.
Während des Trainings wird eine Protein-Datenbank (PDB)-Struktur und ein zufälliger Zeitpunkt (t) ausgewählt, und t-Noising-Schritte werden auf die Struktur angewendet.
RFdiffusion sagt die denoised (pX0) Struktur zu jedem Zeitpunkt voraus, und ein mittlerer quadratischer Fehler wird minimiert zwischen der wahren Struktur (X0) und der Vorhersage (pX0).
Zu den Inferenzzeiten wird eine zufällige Restverteilung (XT) ausgewählt, und RFdiffusion entwirft diese iterativ, um neuartige Proteinstrukturen zu generieren.
Wir haben RFdiffusion hauptsächlich auf Antikörperkomplexstrukturen feinabgestimmt.
Bei jedem Training wird die Antikörperstruktur korrumpiert.
Um die Spezifizierung der Framework-Struktur und -Sequenz zur Inferenzzeit zu ermöglichen, werden die Framework-Sequenz und -struktur als Bedingungsinput während des Trainings an RFdiffusion übergeben.
Da es wünschenswert ist, dass die Rigid-Body-Position (Dock) zwischen Antikörper und Ziel von RFdiffusion zusammen mit den CDR-Schleifen-Konformationen entworfen wird, wird die Framework-Struktur während des Trainings auf eine globalrahmeninvariante Weise bereitgestellt.
Wir nutzen die ‘Template-Track’-Funktion von RF2/RFdiffusion, um die Framework-Struktur als zweidimensionale Matrix von paarweisen Abständen und dihedralen Winkeln zwischen jedem Residuum bereitzustellen.
Die Framework- und Zielvorlagen kodieren nicht ihre relativen Positionen im dreidimensionalen Raum.
In dieser Arbeit haben wir die Sequenz und Struktur der Framework-Region fixiert gehalten und uns auf das Design der CDRs und die allgemeine Rigid-Body-Platzierung des Antikörpers zum Ziel konzentriert.
Wir haben RFdiffusion mit einem zusätzlichen one-hot-kodierten ‘Hotspot’-Feature trainiert, das einen Teil der Rückstände bereitstellt, mit denen die Antikörper-CDRs interagieren, sodass wir bei der Inferenz die Antikörper auf eine bestimmte Stelle ausrichten können.
Mit diesem Trainingsregime ist RFdiffusion in der Lage, Antikörperstrukturen zu entwerfen, die eng mit der Struktur der Eingabe-Framework-Struktur übereinstimmen und das angegebene Epitop mit neuartigen CDR-Schleifen anvisieren.
Nach dem RFdiffusion-Schritt verwenden wir ProteinMPNN, um die CDR-Schleifen-Sequenzen zu entwerfen.
Die entworfenen Antikörper erzielen vielfältige Wechselwirkungen mit dem Ziel-Epitop und unterscheiden sich erheblich von Sequenzen im Trainingsdatensatz.
Es gab keine Korrelation zwischen der Ähnlichkeit des Trainingsdatensatzes und dem Bindungserfolg.
Entwurfspipelines produzieren typischerweise ein breites Spektrum an Lösungen für eine bestimmte Desigaufgabe.
Eine effektive Möglichkeit, entworfene Proteine und Interfaces zu filtern, die wahrscheinlich experimentell erfolgreich sind, basiert auf der Ähnlichkeit der entworfenen Struktur zur von AlphaFold2 vorhergesagten Struktur für die entworfene Sequenz, was gut mit experimentellem Erfolg korreliert.
Im Fall von Antikörpern kann AlphaFold2 jedoch die Antikörper-Antigen-Strukturen nicht genau vorhersagen, was deren Verwendung als Filter in einer Antikörper-Design-Pipeline verhindert.
Zu diesem Zeitpunkt war AlphaFold3 noch nicht verfügbar.
Wir wollten das Designfilter verbessern, indem wir RoseTTAFold2 auf Antikörperstrukturen feinabstimmten.
Um die Strukturvorhersage von Antikörpern zu vereinfachen, haben wir während des Trainings Informationen über die Struktur des Ziels und den Standort des Ziel-Epitops bereitgestellt, an das der Antikörper bindet.
Das feinabgestimmte RF2 muss dennoch die CDRs korrekt modellieren und die richtige Orientierung des Antikörpers zur Zielregion finden.
Der Hintergrund für die Bereitstellung dieser Informationen besteht darin, dass die Zielstruktur und Bindungsposition während des Designs verfügbar sind, jedoch bei allgemeinen Strukturvorhersagen in der Regel nicht.
Mit diesem Trainingsregime und zusätzlichen Informationen ist RF2 in der Lage, wahre Antikörper-Antigen-Paare robust von Scheinpaaren zu unterscheiden und häufig genau Antikörper-Antigen-Komplexstrukturen vorherzusagen, aber nur wenn die gebundene (Holo)-Konformation der Zielstruktur und die Epitope-Informationen bereitgestellt werden.
Bei der Monomer-Vorhersage übertraf das feinabgestimmte RF2 frühere Modelle, die zu diesem Zeitpunkt verfügbar waren, insbesondere hinsichtlich der CDR H3-Strukturvorhersage.
Wenn dieses feinabgestimmte RF2-Netzwerk verwendet wird, um die Struktur von RFdiffusion-entworfenen VHHs neu vorherzusagen, wird ein signifikanter Anteil confident vorhergesagt, an ihrem entworfenen Standort fast identisch zu binden.
Darüber hinaus zeigten In-silico-Cross-Reaktivitätsanalysen, dass RFdiffusion-entworfene VHHs selten an nicht verwandte Proteine gebunden werden.
VHHs, die confident vorhergesagt wurden, an ihrem entworfenen Ziel zu binden, gelten als Hochwert-Interfaces.
Dies deutet darauf hin, dass RF2-Filter wahrscheinlich experimentell erfolgreiche Binder anreichern könnte.
Anfänglich konzentrierten wir uns auf das Design von einzeldomänen Antikörpern (VHHs), die aus Kamelen stammen.
Bis heute wurden zwei VHH-basierte Therapien von der FDA genehmigt, viele klinische Studien sind im Gange.
Obwohl VHHs weniger CDR-Schleifen (drei) als konventionelle Antikörper (sechs) haben, ist die durchschnittliche Wechselfläche eines VHH sehr ähnlich der eines Antikörpers.
Das deutet darauf hin, dass eine Methode, die für das VHH-Design geeignet ist, auch für das Antikörperdesign von Vorteil sein könnte.
In der Tat zeigten In-silico-Metriken für scFvs und VHHs ähnliche Qualitäten von Interfaces, bewertet durch Rosetta und feinstufiges RF2.
Wir wählten ein weit verbreitetes humanisiertes VHH-Framework (h-NbBcII10FGLA) als Grundlage für unsere VHH-Designkampagnen aus.
Wir entwarfen VHHs für eine Reihe von krankheitsrelevanten Zielen: C. difficile TcdB, Influenza H1-Hämagglutinin, Atemwegssyncytialvirus (RSV) Stellen I und III, SARS-CoV-2-Rezeptorbindungsdomäne (RBD) und IL-7Rα.
Computergestütztes Filtern der Designs wurde entweder hochdurchsatzmäßig mit der Hefescreentormethode durchgeführt (9.000 Designs pro Ziel; RSV-Stellen I und III, RBD und Hämagglutinin) oder mit geringerem Durchsatz unter Verwendung von Escherichia coli-Expression und Einzelkonzentration-Oberflächen-Plasmonenresonanz (SPR; 95 Designs pro Ziel; TcdB, IL-7Rα und Hämagglutinin; letzteres wurde mit beiden Methoden getestet).
Die höchsten Affinitätsbinder zu RSV-Stelle III, Influenza-Hämagglutinin, RBD und TcdB sind dargestellt.
Die CDR-Schleifen sind unterschiedlich von VHHs, die in der Natur beobachtet werden, was auf eine erhebliche Generalisierung über den Trainingsdatensatz hinaus hindeutet.
Von den gegen den Insektenzellen-produzierten Hämagglutinin-Monomer getesteten Hämagglutinin-Bindern hatte der am stärksten bindende Binder eine DISS Kd von 78 nM, während andere Binder Affinitäten von 546 nM, 698 nM und 790 nM aufwiesen.
Für TcdB war das Ziel-Epitop die Frizzled-Oberfläche, für die es keine Antikörper oder VHHs im PDB gibt, die diese Stelle anvisieren.
Für den am besten gestalteten VHH sowohl von RBD (Kd = 5,5 μM) als auch TcdB (Kd = 260 nM) wurde die Bindung als an das beabsichtigte Epitop gerichtet bestätigt: Die Bindung wurde vollständig aufgehalten, nachdem ein zuvor entworfener, strukturell charakterisierter de novo-Binder zu diesem Epitop hinzugefügt wurde.
Diese TcdB VHH neutralisierte auch die TcdB-Toxizität in CSPG4-Knockout-Zellen mit einer halben maximalen effektiven Konzentration (EC50) von 460 nM.
Für TcdB waren die Interaktionen spezifisch, wobei keine Bindung zu dem hoch verwandten (70% Sequenz-Homologie) Paeniclostridium sordellii lethaler Toxin L beobachtet wurde.
Diese Daten demonstrieren die Fähigkeit von RFdiffusion, VHHs zu entwerfen, die spezifische Interaktionen mit dem Ziel-Epitop herstellen.
Um die Bindungsaffinität der de novo entworfenen VHHs zu verbessern, nutzten wir das orthogonale fehleranfällige DNA-Replikationssystem, OrthoRep, um eine kontinuierliche Hypermutation der Zielgene in vivo zu ermöglichen.
OrthoRep hat sich als Fähigkeit erwiesen, die schnelle Affinitätsreifung von oberflächen-angezeigten Antikörpern in Hefen voranzutreiben.
Wir haben diese Fähigkeit genutzt, um VHHs anzupeilen, die TcdB, Influenza H1-Hämagglutinin und die SARS-CoV-2 RBD anvisierten.
Affine-maturierte VHHs erwarben verschiedene Mutationen relativ zu den Eltern-Designs und verbesserten die Bindungsaffinitäten um etwa zwei Größenordnungen.
Das macht sie zu geeigneten Kandidaten für die nachfolgende Kryo-EM-Strukturcharakterisierung.
Für TcdB zielte unsere Designkampagne auf das Frizzled-Bindungsepitop ab, das sich auf dem RBD befindet.
TcdB besteht aus vier funktionalen Domänen, einschließlich eines zentralen Delivery- und RBD, an dem die VHHs entworfen werden sollten.
Kryo-EM-Charakterisierung des ursprünglichen Elterndesigns, VHH_TcdB_H2, bestätigte, dass der VHH das Ziel-Epitop des Frizzled DRBD anvisiert.
Die Analyse über zwei dimensionale und dreidimensionale Klassifikation ergab eine Mischung von gebundenen und ungebundenen TcdB-Teilchen.
Ausgezeichnete dreidimensionale Klassifikation und lokale Verfeinerung identifizierten mehrere strukturelle Zustände von TcdB im Datensatz, einschließlich eines erweiterten gebundenen Zustands.
Die dreidimensionale Verfeinerung des gebundenen VHH im erweiterten TcdB-Zustand ergab eine bescheidene Karte mit 4,6 Å, in die das Designmodell zuverlässig starrkörperlich dockte, wobei eine hohe Übereinstimmung mit der beabsichtigten Konstruktstruktur gezeigt wurde.
Um zu bewerten, ob die durch OrthoRep erreichte verbesserte Affinität die ursprüngliche Bindungsart des Elterndesigns beibehielt, führten wir zusätzliche Kryo-EM-Analysen des affinmaturierten VHHs, VHH_TcdB_H2_ortho, durch.
Diese Experimente ergaben einen hohen Anteil von TcdB-Partikeln, die jetzt an den VHH gebunden sind, was mit der erhöhten Affinität übereinstimmte.
Eine ähnliche Verarbeitungs-pipeline wie bei dem Eltern-VHH–TcdB-Komplex ermöglichte es uns, den affinmaturierten VHH–TcdB-Komplex mit einer bescheidenen Auflösung von 5,7 Å zu lösen.
Somit konnten wir den entworfenen VHH zuverlässig in die Kryo-EM-Dichte docken, was bestätigte, dass der VHH das richtige Epitop anvisierte und seine ursprüngliche Bindungsart nach der OrthoRep-gestützten Affinitätsreifung beibehielt.
Diese Ergebnisse unterstreichen die Fähigkeit von RFdiffusion, genaue de novo VHHs zu entwerfen, die in der Lage sind, zuvor unerforschte Epitope gezielt zu erreichen und die für die nachfolgende Affinitätsreifung geeignet sind.
Wir verwendeten Kryo-EM, um einen affinmaturierten VHH (VHH_RBD_D4_ortho19) zu charakterisieren, der das SARS-CoV-2-Spike-RBD anvisierte.
Wettbewerbsexperimente zeigten, dass der Eltern-VHH das beabsichtigte Epitop bindete.
Der RBD wechselt zwischen ‘oben’ und ‘unten’ Konformationen, wobei der ‘obere’ Zustand die Rezeptorbindung und den viralen Eintritt ermöglicht.
Kryo-EM-Zwei-dimensionale Klassifizierungen und dreidimensionale Klassifikationsrekonstruktionen des VHH-gebundenen Komplexes zeigten eine Mischung von RBD-Konformationen (1–2 ‘oben’), wobei VHH-Dichte ausschließlich im oberen Zustand beobachtet wurde.
Dies ist konsistent mit seinem Design, da das Ziel-Epitop in der unteren Konformation verdeckt ist.
Globale Verfeinerung mit einer durchschnittlichen geschätzten Auflösung von 3,9 Å lieferte gut definierte Dichte für den unteren Teil des Spike-Proteins (lokale Auflösung von etwa 2,5 Å), aber die relative Flexibilität des RBD führte zu erheblichem Signalaveraging, was einen Dichteverlust bei höheren Konturwerten zur Folge hatte.
Aus diesem Grund konnte die Genauigkeit des VHH-Designs nicht beurteilt werden.
Symmetrie-Expansion und lokale Verfeinerung halfen, die Auflösung der RBD–VHH-Schnittstelle zu verbessern, und bestätigten die beabsichtigte VHH-Faltung und genaue Epitop-Zieling nach einem starren Docken des Designmodells in die Dichtemap.
Obwohl der VHH das richtige RBD-Epitop anvisierte, wies seine Bindungsart jedoch deutliche Abweichungen vom Designmodell auf, indem er stattdessen eine überwiegend framework-vermittelte Interaktion annahm, die retrospektiv näher mit Vorhersagen von AlphaFold3 übereinstimmte.
Wegen der Abweichung zwischen dem entworfenen Dock und dem experimentell festgelegten Dock klassifizierten wir dies als einen Designfehler.
Angesichts des Erfolgs von RFdiffusion bei der Gestaltung von VHHs mit drei de novo CDRs testeten wir anschließend dessen Fähigkeit, sowohl schwere als auch leichte Ketten im scFv-Format zu entwerfen.
RFdiffusion wurde verwendet, um scFvs zu generieren, die spezifische Epitope anvisieren, wobei eine ähnliche Strategie wie beim VHH-Design verfolgt wurde.
Im Gegensatz zu VHHs, bei denen nur drei CDRs de novo erstellt wurden, beinhaltete das scFv-Design den Aufbau aller sechs CDRs sowohl auf den schweren als auch auf den leichten Ketten zusätzlich zur Docking-Modus.
Das Gene-Syntheseproblem ist für scFvs herausfordernder als für VHHs, da sie zu lang sind, um einfach aus Paaren konventioneller Oligonukleotide, die auf Oligonukleotidarrays synthetisiert wurden, assembling zu werden.
Die spezifische Paarung ist aufgrund der hohen Sequenzhomologie zwischen scFvs schwierig.
Wir entwickelten schrittweise Assemblierungsprotokolle, die es ermöglichen, Bibliotheken mit schweren und leichten Ketten sowohl spezifisch gepaart wie in den Entwurfsmodellen als auch kombinatorisch gemischt innerhalb von Design-Untergruppen mit ähnlichen Zielbindemodi zu konstruieren.
Dieser Ansatz hilft, die größere Herausforderung der genauen Gestaltung von sechs CDRs de novo zu überwinden, was die Möglichkeiten für Fehler erhöht, da bereits ein suboptimales CDR das Binding kompromittieren kann.
Wir stellten fest, dass gegebenen Sets von nahezu superimponierbaren Designs, die auf die gleiche Stelle mit dem gleichen Bindungsstil abzielen, neue scFvs, die durch das Kombinieren von Paaren aus schweren und leichten Ketten aus verschiedenen Designs generiert wurden, confident vorhergesagt wurden, um an die Zielstelle mit ähnlichen Frequenzen zu binden wie die ursprünglichen Designs.
Im Gegensatz dazu führte die zufällige, strukturagnostische Paarung selten zu vorhergesagten Bindern.
Somit können wir durch das Mischen von CDRs aus verschiedenen Designs, die in derselben Ausrichtung binden, effektiv Fehlschläge aufgrund einzelner, ungenau gestalteter CDRs überwinden, was somit eine kombinatorische Lösung für ein kombinatorisch komplexeres Problem darstellt (Zweiketten-scFv-Design vs. Einketten-VHH-Design).
Diese Strategie hebt einen wichtigen Vorteil des strukturbasierenden Designs hervor: die ‘intelligente’ Paarung von schweren und leichten Ketten ist mit einem strukturellen Modell jedes Antikörpers möglich und ermöglicht es, de novo entworfene Antikörperbibliotheken, die in den Maßstäben erreicht werden, die durch traditionelle Bibliotheksversammlungs-Methoden jedoch zurzeit aktuell in der Gen-Synthese vorhanden sind.
Wir waren erfolgreich, epitopspezifische scFvs aus den schweren-leichten kombinatorischen Bibliotheken (mit einer theoretischen Komplexität von ungefähr 10 Millionen) zu identifizieren, jedoch nicht aus den festen Pairing-Bibliotheken.
Nach der Expression und Reinigung zeigte die SPR-Analyse von sechs distinct scFvs, die aus zwei einzigartigen Docks stammen, welche das Frizzled-Epitop von TcdB anvisieren, eine Vielzahl von Affinitäten.
Der am höchsten affine Binder, scFv6, hatte einen Kd von 72 nM.
Die Umwandlung des scFv in ein vollständiges IgG1 erzeugte Antikörper, die mit vergleichbarer (68 nM) Affinität binden, was zeigt, dass unsere Desigmethode zur Generierung von vollständigen Antikörpern verwendet werden kann.
Es gibt keine Antikörper, die an dieses Epitop im PDB binden, sodass dieser Erfolg nicht auf Memorierung zurückzuführen ist.
Die nachfolgende Vorhersage der Struktur des scFv mit AlphaFold3 zeigte eine Bindungsart, die identisch war mit der der beiden nahezu superimponierbaren Herkunftsdesigns, die die leichten und schweren Ketten beitrugen.
Wettbewerb mit einem bekannten Rezeptor, Frizzled-7, zu diesem Epitop bestätigte, dass das binden von scFv5 zielgerichtet war.
Im Gegensatz dazu wurde im Beisein von CSPG4, einem alternativen Rezeptor, der mit einem Epitop im Toxin-Kern interagiert, keine Konkurrenz beobachtet.
Das zeigt, dass scFvs aus strukturbewussten, designten kombinatorischen Bibliotheken identifiziert werden können.
Wir zielten anschließend auf ein klinisch relevantes Epitop ab: das QYNPIRTTF-Peptid, das vom neuroblastomabhängigen PHOX2B-Gen und Mastertranskriptionsregulator in Komplex mit dem Haupt-Histokompatibilitätskomplex (MHC) Allotyp HLA-C*07:02 abgeleitet wurde.
Das PHOX2B-Peptid wurde ursprünglich durch Immunpeptidomik von patientenabgeleiteten Neuroblastomproben entdeckt und wurde mit Peptid-zentrierten chimerischen Antigenrezeptoren (PC-CARs) zur Behandlung von Hochrisiko-Neuroblastom angesprochen.
Jedoch sind die zuvor identifizierten PC-CARs darauf beschränkt, PHOX2B zu erkennen, der auf HLAs der A9-serologischen Gruppe präsentiert wird, wobei der verbreitete Allotyp HLA-C*07:02 ausgeschlossen wird.
Das Targeting des PHOX2B–HLA-C*07:02-Komplex kann potenziell die adressierbare Patientenzahl für diese Immuntherapien signifikant erhöhen und war Gegenstand der laufenden Therapeutik-Entwicklung.
Kürzlich wurden computergestützt entworfene (nicht-Antikörper) Binder für PHOX2B–HLA-C*07:02 entwickelt, unter Verwendung des TRACeR-I-Systems, während hochaffine TCRs zur Zielerfassung von Peptiden auf den gemeinsamen HLA-C*08:02/HLA-C*05:01-Allotypen identifiziert wurden.
Ein Vorteil des strukturzentrierten Designs besteht darin, dass es auf spezielle Peptidreste abzielt, um Bindungsspezifität zu erreichen (anstatt nur an das MHC zu binden), weshalb wir RFdiffusion einsetzten, um den R6-Rest anzusprechen, der bekannt ist, dass er für die Bindung beim PC-CAR wichtig ist.
Angesichts der geringen Stabilität des PHOX2B–HLA-C*07:02-Komplexes (Tm von 44,2 °C) haben wir einen disulfid-stabilisierten Ansatz genutzt, um eine stabilisierte Form des pHLA-Ziels vorzubereiten.
Durch Verwendung des oben beschriebenen kombinatorischen Assemblierungsansatzes identifizierten wir bescheidene Affinitäts-scFv-Binder zu PHOX2B-HLA-C*07:02.
Die Bindung war spezifisch für das Peptid, wobei keine nachweisbare Bindung an das R6A-Punktmutant PHOX2B-Peptid (PHOX2B(R6A)–HLA-C*07:02) beobachtet wurde.
Es wurden Versuche unternommen, scFv-Binder in einen 4-1BB-CAR zu integrieren, jedoch zeigten T-Zell-Zytotoxizitätstests, dass keine nachweisbare Tötung einer Reihe von Neuroblastomzelllinien stattfand.
Das ist wahrscheinlich auf die bescheidene Bindungsaffinität und oder geringe Antigendichte zurückzuführen, die auf den Tumorzellen exprimiert werden.
Obwohl es noch erheblichen Spielraum zur Verbesserung der Affinität gibt, demonstriert dies die Fähigkeit des strukturzentrierten Antikörperdesigns, zusammen mit geeigneten Bibliotheksversammlungsverfahren spezielle Binder an herausfordernden und klinisch wichtigen Ziel-Epitopen zu entwerfen.
Um die Genauigkeit des de novo scFv-Designs zu bewerten, bestimmten wir die Kryo-EM-Strukturen von zwei kombinatorisch montierten scFvs, scFv5 und scFv6, die beide das Frizzled-Epitop von TcdB anvisieren.
Die Kryo-EM-Analyse bestätigte, dass beide scFvs das Frizzled-Epitop wie entworfen bindeten.
Hochauflösende zweidimensionale Klassifizierungen von scFv6 zeigten klare Dichte für sowohl TcdB als auch für den gebundenen scFv, untermauert von einer drei-dimensionalen Rekonstruktion mit 3,6 Å.
Die aufgelöste Struktur zeigte, dass scFv6 das Frizzled-Epitop entlang seiner DRBD-Domäne mit der vorhergesagten Bindungsorientierung ansprach.
Superposition der Kryo-EM-Struktur mit dem Designmodell zeigte bemerkenswerte Übereinstimmung, wobei sowohl schwere als auch leichte Ketten mit dem Epitop interagierten wie beabsichtigt.
Die gesamte Faltung passenden dem Design eng, einem Kompositmodells der beiden Ketten, das aus unterschiedlichen, aber strukturell ähnlichen Designs stammt, zeigt einen RMSD von 0,9 Å.
Jede der sechs CDRs zeigte nahezu atomare Präzision (Rückgrat-RMSDs: CDRH1 = 0,4 Å, CDRH2 = 0,3 Å, CDRH3 = 0,7 Å, CDRL1 = 0,2 Å, CDRL2 = 1,1 Å und CDRL3 = 0,2 Å).
Diese Übereinstimmung erstreckte sich auf die Rotamer-Konformationen der CDR-Seitenketten und deren Wechselwirkungen mit dem Frizzled-Epitop, was die Genauigkeit von RFdiffusion im Design von de novo CDR-zielgerichteten Interaktionen unterstreicht.
ScFv5 wurde zur Bindung an dasselbe Epitop, jedoch mit einem unterschiedlichen Annäherungswinkel gegenüber scFv6 entworfen.
Eine 6,1 Å Kryo-EM-Rekonstruktion bestätigte die Bindung von scFv5 an das TcdB Frizzled-Epitop, wobei zweidimensionale Klassifikationen klar Dichte für den Komplex zeigten.
Ein starres Docken des Designmodells in die Kryo-EM-Dichte offenbarte eine enge Übereinstimmung zwischen den Vorhersagen der Bindungs- und den experimentellen Bindungsmoden.
Obwohl unsere Ergebnisse zeigen, dass das de novo Design von Antikörperdomänen möglich ist, bleiben die experimentellen Erfolgsraten gering.
Ein entscheidender Faktor für frühere Erfolge beim de novo Binderdesign war die Verbesserung der Filter (hauptsächlich AlphaFold2), welche Experimentellen Erfolg in dem Teil des Designs, der getestet wird anreicherte.
Zu Beginn dieser Studie strebten wir an, ein solches Filter zu erstellen, indem wir RoseTTAFold2 verfeinerten.
Jedoch ist die Filterkraft dieses Modells begrenzt.
Dies erklärt wahrscheinlich die niedrigen experimentellen Erfolgsraten und die ungenaue SARS-CoV-2 design, in dem die Gesamtfaltung und die Zielbestimmung korrekt waren, jedoch die Bindungsorientierung nicht.
Nach der Designarbeit in dieser Studie wurde AlphaFold3 veröffentlicht, wodurch die Genauigkeit der Antikörperstrukturvorhersage verbessert wurde.
Im Nachhinein können wir bewerten, wie das Filtern mit AlphaFold3 die experimentellen Erfolgsquoten erhöht hätte.
Erstens sagt AlphaFold3 die experimentell validierte Struktur des ungenau entworfenen SARS-CoV-2 VHH genau voraus.
Hätte AlphaFold3 als anfängliches Filter verwendet werden, hätte dieses Design aufgrund der Diskrepanz zwischen den vorhergesagten und beabsichtigten Strukturen verworfen werden müssen, wodurch die experimentelle Prüfung verhindert worden wäre.
Zweitens sagten wir die Strukturen von SARS-CoV-2, Influenza-Hämagglutinin, TcdB und IL-7Rα VHH-Designs mit AlphaFold3 vorher, wobei wir ein multiples Sequenz-Alignment (MSA) und Vorlagen für das Ziel und nur eine Vorlage für die VHH verwendeten.
Wir analysierten die Vorhersagen für Bibliotheken mit zumindest einem strukturell validierten VHH.
Die Ergebnisse werden stark von anti-Hämagglutinin-VHHs dominiert, da die meisten erfolgreichen Binder aus dieser Bibliothek stammen.
Wir fanden heraus, dass der AlphaFold3-Schnittstellenvorhersage-Template-Modell-Score, ein Maß für das Vertrauen im Modell über die Schnittstelle, prädiktiv für den Bindungserfolg ist.
Insgesamt haben nur 9 % unserer bestellten VHH-Designs einen ipTM > 0,6, was darauf hindeutet, dass die Erfolgsquote durch die Einbeziehung eines ipTM-Filters erhöht wird.
Wir führten eine ähnliche Analyse für die kombinatorisch montierten scFv-Bibliotheken durch; wir sagten die Strukturen der Eltern-scFv-Entwürfe (vor kombinatorischer Assemblierung) und die experimentell bestätigten scFv-Designs (kombinatorisch montiert) mit AlphaFold3 unter Verwendung eines MSA für die Zielsequenz und Vorlagen für das Ziel sowie die schweren und leichten Ketten voraus.
Wir nahmen den maximalen ipTM-Score über 10 Seeds.
Wir fanden heraus, dass erfolgreiche Designs sich zu höheren AlphaFold3 ipTM Scores clustern als die Eltern-Designs.
Nur 4 % der anfänglichen Designbibliothek haben ipTM > 0,85, während 5 von den 6 experimentell bestätigten Designs diese Schwelle überschreiten, was erneut darauf hindeutet, dass das Filtern durch AlphaFold3 ipTM die Erfolgsquoten erhöhen sollte.
Unsere Ergebnisse zeigen, dass das de novo Design von Antikörperdomänen möglich ist, die spezifische Epitope auf einem Ziel anvisieren.
Die Kryo-EM-Strukturdaten für die entworfenen VHHs zu Influenza-Hämagglutinin und TcdB zeigen eine sehr enge Übereinstimmung mit den computergestützten Designmodellen, was zeigt, dass unser Ansatz VHH-Komplexe mit atomarer Genauigkeit gestalten kann, einschließlich der hochvariablen H3-Schleife und der allgemeinen Bindungsorientierung, die hochunterschiedlich mit allen bekannten Strukturen im PDB sind.
Darüber hinaus zeigen Kryo-EM-Strukturdaten von entworfenen scFvs, die an TcdB gebunden sind, die Fähigkeit von RFdiffusion, präzise zwei-Ketten-scFvs zu entwerfen.
Soweit uns bekannt ist, sind dies die ersten strukturell validierten Beispiele für de novo entworfene Antikörper.
Unsere computergestützte Methode synergiert mit experimentellen Screening-Methoden, die für die Rückgewinnung von Antikörpern aus großen zufälligen Bibliotheken entwickelt wurden, auf mehrere Weise.
Erstens ermöglichen Hefedisplay-Auswahlmethoden, die weit verbreitet für das Screening von Antikörperbibliotheken verwendet werden, die Rückgewinnung der bindenden Bindehöhen unter großen Design-Sets, was derzeit aufgrund der relativ niedrigen Design-Erfolgsquote notwendig ist.
Zweitens ermöglicht das Screening von kombinatorischen Bibliotheken, die schwere und leichte Ketten aus Designs mit ähnlichen Bindemodi mischen, die Identifizierung von scFvs, die aus strukturell kompatiblen Ketten bestehen, die spezifische Epitope anvisieren, wie hier für TcdB und PHOX2B–Peptid-MHC gezeigt wurde.
Drittens verbessert die Affinitätsreifung unter Verwendung von OrthoRep die gemessene Affinität der ursprünglichen VHH-Designs auf den Nanomolar- oder Subnanomolarbereich, während sie die ursprüngliche beabsichtigte Bindungsart beibehält.
Aus praktischer Sicht besteht der entscheidende Fortschritt dieser Arbeit nicht darin, VHHs und scFvs gegen ein Ziel zu generieren – etwas, das oft durch reine experimentelle Methoden erreichbar ist –, sondern vielmehr darin, genau spezifische Bindungs-Epitopen zu zielen.
Die Epitopspezifität ist entscheidend für therapeutische Anwendungen wie Antagonisten, die Rezeptor-Ligand-Interaktionen blockieren, Antikörper, die vermeiden, mit endogenen Molekülen zu konkurrieren, Modulatoren, die konformationale Veränderungen auslösen, um die Signalgebung zu triggern oder Antikörper targeting auf konservierte oder evolutionär eingeschränkte virale Epitopen.
Es gibt noch erheblichen Verbesserungsspielraum.
Für den Rückgrat-Design-Schritt könnte die Einbeziehung jüngster architektonischer Verbesserungen und neuer Fortschritte im generativen Modellierung höhere Designfähigkeit und Diversität erbringen.
RoseTTAFold2 und RFdiffusion wurden auch kürzlich erweitert, um alle Biomoleküle zu modellieren.
Die Integration dieser Fähigkeit in die Antikörper-Design-RFdiffusion-Variante sollte das genaue Design von Antikörpern für Epitopenhantierungen, die nicht-aus-Protein-Atome enthalten, wie Glykane, ermöglichen.
ProteinMPNN wurde in diesem aktuellen Werk nicht modifiziert, jedoch wird das Design von Sequenzen, die enger an den menschlichen CDR-Sequenzen liegen, voraussichtlich die potenzielle Immunogenität der entworfenen Antikörper verringern.
In der Tat sind entworfene Sequenzen derzeit etwas weniger menschlich than therapeutische Antikörper CDRs.
Zusätzliche Verbesserungen der Vorhersagemethoden für Antikörperstrukturen sollten eine schnellere Optimierung der vorgelagerten Entwurfsmethoden und verbesserte experimentelle Erfolgsquoten ermöglichen.
Schließlich könnte das computergestützte de novo Design von Antikörpern, unter Einsatz unserer RFdiffusion- und verwandten Ansätzen, die Antikörperentdeckung und -entwicklung revolutionieren.
Mit der Verbesserung der Methode und den steigenden Erfolgsraten hat sie das Potenzial, schneller und kostengünstiger zu sein als die Immunisierung von Tieren oder das Screening zufälliger Bibliotheken.
Ein strukturzentrierter Ansatz zum Antikörperdesign sollte auch die Optimierung wichtiger pharmazeutischer Eigenschaften wie Aggregation, Löslichkeit und Expressionsebenen unterstützen, die alle bedeutende Herausforderungen in der Antikörperentwicklung darstellen.
Zusammen erwarten wir, dass das computergestützte Design von Antikörpern die Anzahl der ansprechbaren klinischen Targets und Krankheiten, die für Antikörpertherapeutika zugänglich sind, erhöhen wird.
Bildquelle:nature